Theoretical Background
Algorithms
BioXL/H algorithms utilize the dynamic programming method of sequence alignment by Needleman & Wuncsh (Journal of Molecular Biology, 48, 443-453, 1970), which was adapted for optimal local alignments by Smith & Waterman (Journal of Molecular Biology, 147, 195-197, 1981), with modifications by Gotoh (Journal of Molecular Biology,162, 705-708, 1982).
In Smith-Waterman database searches, BioXL/H uses the dynamic programming method to compare every database sequence to the query sequence and assign a score to each result. ProfileSearch, developed by Gribskov, et. al. (Proc. Natl. Acad. Sc. USA 84: 4355-4358, 1987), uses the same dynamic programming methodology; however, the query is a profile created from the alignment of multiple sequences.
HMMSearch, developed by Sean Eddy, et. al. (Dept. of Genetics, Washington University School of Medicine, HMMER package, http://genome.wustl.edu/eddy/hmmer.html), uses a more comprehensive profile structure and dynamic programming methodology to produce more sensitive homology measurement on the amino-acid level.
Dynamic Programming Model
The dynamic programming method checks every possible alignment between two given sequences. The two sequences define a matrix in which every cell represents the alignment of two specific positions in the two sequences. The alignments between specific positions are given either positive or negative scores, depending on the residues located in these positions.
The score of alignment in a certain position is then added to the sum of the highest scores of alignment leading to that position. The highest score can sometimes be achieved by adding gaps to the alignment, although introduction of a gap is penalized. After all the cells have been calculated, the position of the highest score is the final aligned position of the best local alignment between the two sequences.
In Figure 4, a sample matrix shows the alignment of sequence X (CAGCCUCGCUUAG, from left to right on the horizontal axis) and Y (AAUGCCAUUGACGG, from top to bottom on the vertical axis). The matrix is taken from Smith & Waterman (Advances in Applied Mathematics, 2, 482-489, 1981).
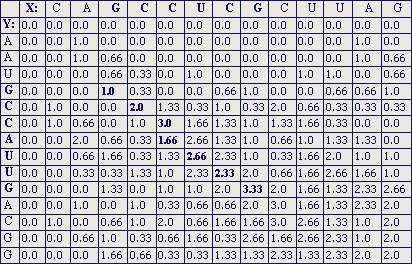
:: Figure 4: Sample Matrix
Scores in the first row and column are defined as zero. Entries Hi,j in all other cells of the matrix are defined as the score of the best alignment ending in the position matching xi and yj, and are calculated using the following equation (from Waterman and Vingron, Proc. Natl. Acad. Sci. USA, 91, 4625, 1994):
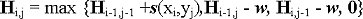
where
s(x i ,y j ) is the score of a match between xi and yj specified in the scoring matrix w is the gap penalty, which is a function of two parameters. If a new gap is opened, w equals (g+k), where g is a penalty for opening a gap, and k is a penalty for extending a gap. If a previously opened gap is extended, w equals k. The total penalty for a gap of length l is g + k*l. This is an "affine scoring penalty," as opposed to a non-affine penalty, for which g is zero.
H i,j equals to the first term in the equation if the maximum score is generated by aligning x i and y j . H i,j equals to the second term if a gap in the sequence Y is extended or opened in the current position, in which case the gap penalty is added to the score in the cell to the left of the current cell. H i,j equals to the third term if a gap in the sequence X is extended or opened in the current position, in which case the gap penalty is added to the score in the cell above the current cell.
Calculation of the best local alignment can therefore be viewed as diagonal movement from the top left cell to the bottom right cell. After scores in all cells are calculated, the highest score in the matrix is located in the final position of the best local alignment.
The sample matrix in Figure 4 was generated using a scoring matrix that assigns 1 to identical matches, and -1/3 to mismatches. The gap open penalty is 1 and the gap extend penalty is 1/3. The alignment leading to the cell with score 3.33 is the best local alignment:
Y: G C C A U U G
X: G C C . U C G
There is one gap in the alignment (represented as "." in sequence X), and one mismatch in the sixth position.
Speed of Calculations
Because calculation of the best local alignment requires calculation of scores in all cells in the matrix, the speed of computing the best alignment can be measured in matrix cell updates per second (MCPS). Software implementations of the Smith-Waterman algorithm, such as the BestFit application from GCG, can run at speeds of about thirty million MCPS on high-end workstations.
BioXL/H-8 can run at approximately six billion MCPS for long sequence queries. A search of all GenBank sequences (the total of 10 billion base pairs) with a DNA sequence of 1000 bases in length requires calculation of (1000*2*10,000 million) = 20,000 billion cells. The factor of 2 is added because both the sequence and its complement strand are searched. On BioXL/H-8, this search would take about 3300 seconds (about 56 minutes). The same search will take 11,100 minutes (more than seven and a half days) on a high-end UNIX machine.
|